Glioblastoma carries poor prognosis, with five-year survival of just 6.8% despite surgery, chemotherapy, and radiation (RT). New therapies are urgently needed. Tumor metabolism in GBM is critical for accelerated growth because of upregulation of glucose, amino acid, and fatty acid utilization. Unfortunately, therapies targeting GBM metabolism, whether with small-molecule compounds or dietary interventions to limit nutrient sources, have failed in clinical trials due to induction of tumor escape pathways. Existing therapies (RT and temozolomide, TMZ) indirectly target metabolism by inhibiting the redox buffering capacity in GBM cells, which is required during nutrient oxidation. It is unknown if combination strategies that target metabolism will synergize to improve treatment efficacy and avoid tumor escape.
Image
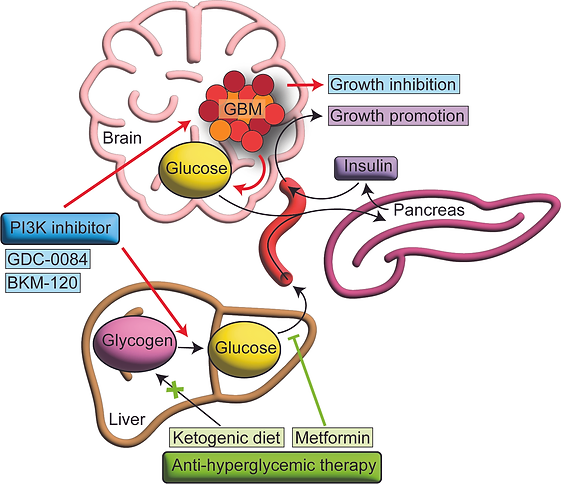
We first identified a strategy to more effectively target the phosphatidylinositol 3-kinase pathway, which is mutated or activated in 90% of patients with GBM, by preventing insulin feedback. The efficacy of PI3K inhibition for cancer treatment is impaired by on-target insulin feedback that reactivates the PI3K signaling pathway. Treatment of patients with recurrent GBM using PI3K inhibitors failed in a Phase 2 clinical trial. We demonstrated that PI3K inhibitors induce hyperglycemia and hyperinsulinemia in mice and that counteracting hyperglycemia with the anti-hyperglycemic drug metformin enhances therapeutic efficacy in an orthotopic GBM mouse model. We also found that hyperglycemia was an independent factor associated with poor progression-free survival in a clinical trial evaluating PI3K inhibition in patients with recurrent GBM. This hyperglycemia was sufficient to induce insulin receptor activation in tumor tissue from patients. Implementation of a rational anti-hyperglycemia regimen along with PI3K inhibition may help translate these findings to improve PI3K targeting in patients with GBM.
Image
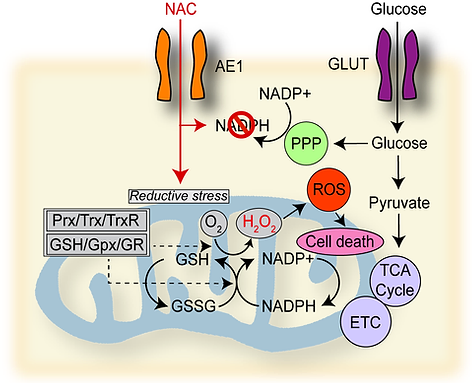
Our recent work involves our discovery of a metabolic susceptibility to cysteine-promoting molecules in GBM. Surprisingly, we found that treatment of patient-derived GBM cells with the FDA-approved cysteine compound N-acetylcysteine (NAC), which is a well-known antioxidant, reduced GBM cell growth and mitochondrial respiration. This effect was exacerbated by glucose starvation. Normal brain cells and other cancer cells showed no response to NAC. Mechanistic experiments revealed that cysteine compounds induce rapid mitochondrial hydrogen peroxide production and reductive stress in GBM cells, an effect blocked by oxidized glutathione, thioredoxin, and redox enzyme overexpression. Analyses of public genomic and proteomic databases identified that GBM cells exhibit the highest level of cysteine-metabolic genes of 32 cancers and lower expression of mitochondrial redox enzymes than 4 other cancers whose proteomic data is available. We found that mitochondrial redox enzyme deficiency underlies this effect and can be exploited therapeutically in GBM by exposing mice bearing orthotropic GBM xenografts to cysteine-promoting peptides. These findings indicate that GBM is uniquely susceptible to NAC-driven reductive stress and could synergize with glucose-lowering treatments for GBM.
We are working on experiments to identify the effect of omega-3 and omega-6 fatty acids on GBM cell growth and lipid metabolism, with the goal of translating these findings into dietary studies in patients.