Our Mission
The mission of our laboratory is to contribute to the development of therapies and a cure for Friedreich’s ataxia (FRDA) by elucidating molecular mechanisms causing the disease, developing novel cellular and animal models of FRDA, identifying disease biomarkers, and testing novel therapeutic approaches.
Friedreich’s Ataxia
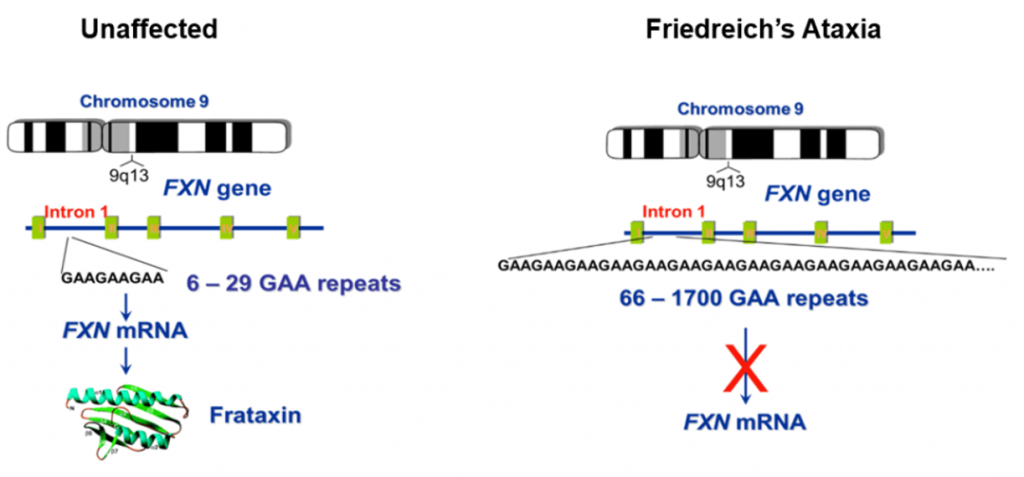
Our laboratory is focused on Friedreich’s ataxia, the most inherited ataxia in humans. The most common mutation causing approximately 95% of the cases of FRDA is an expansion of a trinucleotide repeat sequence – GAA – located in intron 1 of the frataxin (FXN) gene. Unaffected alleles contain up to approximately 40 GAA repeats, while disease alleles are larger than 70 triplets, with most spanning 600 – 900 GAA units and frequently exceeding 1,000 repeats.
In the unaffected population, the GAA repeat tracts in FXN are short and stable (i.e., the number of GAA units does not change in different cells or tissues, or during germline transmission). However, in FRDA individuals, the expanded repeat tracts become unstable and the number of GAAs differs amongst tissues and changes during transmission to the next generation. Interestingly, approximately 1 in 100 individuals in the population carry one expanded GAA tract. These mutation carriers are asymptomatic.
The expanded GAA repeats alter the synthesis of FXN mRNA, triggering local chromatin changes in the FXN gene, resulting in transcriptional silencing. FXN levels in patient samples are reduced by 70 – 95%. The lengths of the expanded GAA tracts determine the extent of FXN downregulation, and FXN levels correlate inversely with the age of disease onset and directly with disease severity and progression.
Approximately 5% of FRDA patients are compound heterozygous with one FXN allele harboring expanded GAAs and the second allele containing a point mutation. Importantly, from a therapeutic point of view, all FRDA patients produce some, albeit a limited amount of, functional frataxin. Frataxin participates in iron-sulfur cluster (ISC) biosynthesis in the mitochondria, and many of the overt FRDA phenotypes result directly from deficient activity of proteins containing ISC cofactors, such as mitochondrial respiratory complexes, Krebs cycle proteins, DNA repair, and replication proteins.
Generally, FRDA model systems demonstrate disturbed intracellular iron homeostasis. Ubiquitous functions of ISCs as prosthetic groups in a variety of critical pathways result in the pleiotropic and multi-organ manifestation of FRDA.
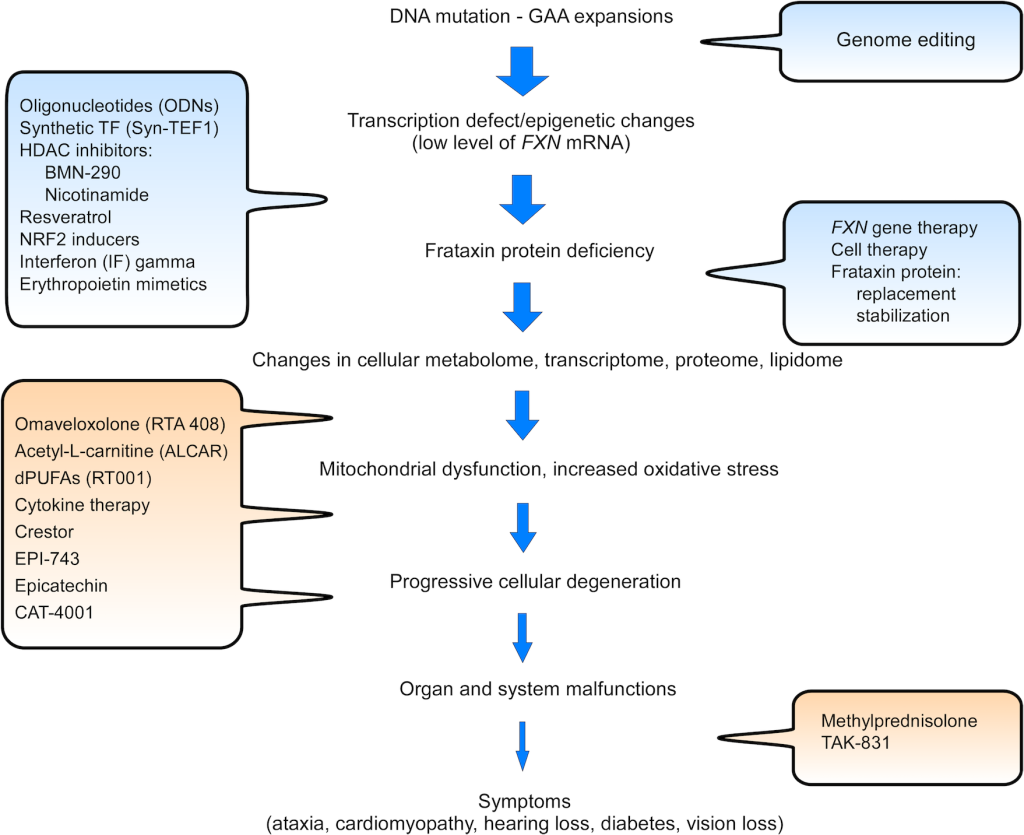
Currently, there is no cure for FRDA, and research efforts are focused on two general strategies:
- To increase FXN levels via stimulation of endogenous gene expression or by gene/protein replacement
- To treat downstream consequences of FXN deficiency via targeting increased production of reactive oxygen species (ROS), mitochondrial dysfunctions, etc.
A summary of major therapeutic initiatives is shown in the figure below. A current therapeutic pipeline can also be found on Friedreich’s Ataxia Research Alliance (FARA) website.
PMID: 30905359
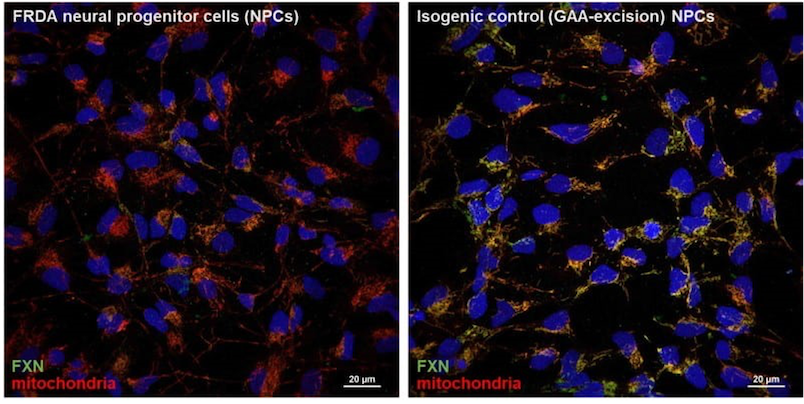
We conduct our studies in different FRDA cellular model systems starting with FRDA fibroblasts and iPSCs, then in neurons and cardiomyocytes differentiated from iPSCs. We also use genome editing techniques (such as CRISPR-Cas9) to excise the expanded GAA repeats from FRDA cells, creating isogenic control lines like the example shown below. Finally, we developed two novel mouse models of FRDA for studies requiring in vivo approaches.
Our laboratory takes a multidisciplinary approach to study the molecular mechanisms underlying Friedreich’s ataxia. Current projects include:
- Elucidating the mechanism of transcription silencing induced by expanded GAA repeats in the FXN gene
- Developing therapeutic strategies aimed at reactivating transcription of the FXN gene, including antisense oligonucleotides (ASOs), transcription activators, and gene therapy
- Utilizing genome editing as a regenerative therapy approach to FRDA
- Defining mitochondrial changes in FRDA cells
- Identifying molecular biomarkers of FRDA
- Characterizing molecular phenotypes of FRDA neuronal and cardiac cells derived from induced pluripotent stem cells (iPSCs)
- Defining effects of point mutations on frataxin functions and their resulting disease phenotypes
- Generating novel cellular and mouse models of FRDA
Contact Us
The Napierala Lab
NL9.110
(214) 648-4279
Marek Napierala, Ph.D.
UT Southwestern Medical Center
5323 Harry Hines Blvd.
Suite NL9.108
Dallas, TX 75390-8813
(214) 648-3551
Email
Jill S. Napierala, Ph.D.
UT Southwestern Medical Center
5323 Harry Hines Blvd.
Suite NL9.108
Dallas, TX 75390-8813
(214) 648-8266
Email
Shipping Address:
Dr. Marek Napierala
UT Southwestern Medical Center
5901 Forest Park Rd.
Suite NL9.110
Dallas, TX 75390-8813
(214) 648-3551