 LDL Receptor Gene Family
LDL Receptor Gene Family Alzheimer's disease and atherosclerosis are progressive degenerative syndromes that together afflict more than 50% of the population in Western societies. Their prevalence and socioeconomic impact are steadily increasing along with average life expectancy. Our work has identified several fundamental molecular mechanisms that are common to both of these superficially unrelated diseases. They are mediated by a class of ancient and evolutionarily highly conserved cell surface receptors known as the LDL receptor gene family.
The core of this family consists of seven structurally closely related multifunctional receptors that share partly overlapping roles in such diverse biological processes as receptor-mediated endocytosis, regulation of extracellular protease activity, hormone transport, and intercellular signaling. The range of physiological functions in which the receptors are involved includes lipid metabolism, neuronal migration during brain development, neurotransmission, axonal transport, vitamin metabolism, and the control of cellular proliferation. All members of the family are also receptors for Apolipoprotein E (ApoE), a major risk factor gene for late-onset Alzheimer’s disease. One of our goals is to understand how the interaction of ApoE and other physiological ligands with its receptors affects the cell biology of the neuron.
Our group uses complex conventional and conditional gene targeting and transgenic approaches in mice to investigate the molecular basis by which LDL receptor family members control pivotal mechanisms of cellular communication during embryonic development and in the adult organism. We employ numerous strains of mice with compound mutations in multiple genes to investigate 1) the role of the ApoE receptor-2 and the VLDL receptor and their ligand Reelin in neuronal migration, axonal transport, neurotransmission, and neurodegeneration, 2) a conceptionally novel therapeutic approach to target ApoE4 carriers through the acidification of the early endosomes, 3) the role of progranulin in frontotemporal dementia, 4) a novel approach in targeting Reelin in inflammatory diseases, and 5) the role of LDL receptor-related protein (LRP) in the regulation of smooth muscle cell signaling, vascular remodeling, and integrity in atherosclerosis.
Reelin - a novel target for the prevention of inflammatory disease.
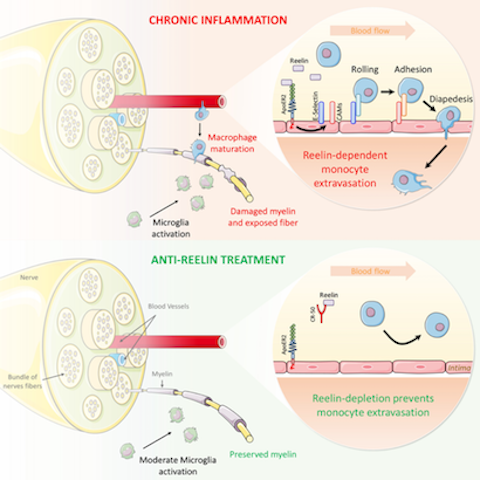 Anti-Reelin Treatment for Chronic Inflammation
Anti-Reelin Treatment for Chronic Inflammation Under normal conditions, the endothelial barrier effectively regulates the passage of immune cells. However, under chronic inflammatory conditions like multiple sclerosis (MS), leukocytes, especially monocytes, infiltrate the tissues (CNS) where they promote inflammatory demyelination, resulting in paralysis. Therefore, all current therapeutic approaches to MS target the immune system, blocking inflammation and paralysis progression. We have developed a mechanistically different and unique therapeutic approach selectively targeting endothelial permeability to block neuroinflammation and MS progression by preventing monocyte extravasation to the central nervous system. Our three major results describe: 1) A novel pathophysiologic function: initially recognized only for guiding neurons during brain development, we propose a novel non-neuronal function for Reelin which “guides” leukocytes by increasing vascular permeability, thus paving the way for leukocyte extravasation. 2) A non-cytokine biomarker: circulating Reelin is increased during MS relapse and normalized to control in remission patients, thus associated with MS state and severity. 3) An original non-immunosuppressive approach for chronic inflammation: in an MS mouse model, anti-Reelin treatments reduce monocyte extravasation, preventing neuroinflammation, demyelination, and ultimately paralysis progression.
By expanding our understanding of the role Reelin may play in MS pathogenesis, this translational project may incite the development of therapeutic strategies not only for MS but also for other chronic inflammatory diseases like atherosclerosis, arthritis, or Crohn’s disease, which positively impact patient’s quality of life.
Reelin depletion protects from multiple sclerosis by reducing monocyte extravasation and inflammation.
This mechanistic model incorporates the findings described in this article and the literature to date, as discussed. Reelin promotes the expression of vascular adhesion proteins, thereby increasing monocyte adhesion/extravasation, inflammation, and consequently demyelination. Both genetic and therapeutic Reelin depletion prevents this inflammatory cascade and thus paralysis in a murine EAE model. We surmise that the same mechanism is conserved in humans where Reelin also promotes monocyte adhesion to vascular endothelial cells in vitro.
Featured in ScienceDaily.
NHE6: A Rational Target to Prevent Alzheimer’s Disease
It has been a century since Alzheimer’s disease (AD) has been described and despite an enormous amount of research into the pathogenesis of AD and therapeutic strategies including, but not limited to, inhibitors of β-secretase, immunotherapy against Aβ or tau, and cholinesterase inhibitors no effective treatment has so far materialized. The core mission of my Center for Translational Neurodegeneration Research (established in 2014) is to identify rational therapeutic strategies for preventing or delaying neurodegenerative diseases including AD. To achieve this goal, our laboratories investigate their pathophysiology and molecular mechanisms, which informs us of the most promising avenues for effective drug development, followed by the preclinical and clinical evaluation of their efficacy.
Approximately 95% of AD patients develop late-onset AD (LOAD). The most important risk factor for LOAD is the Apolipoprotein E (ApoE) isoform ε4 genotype. The reason why ApoE4 increases the AD risk is still being vigorously debated. My laboratory has discovered and was the first to show that ApoE4 induces a defect in endolysosomal trafficking or the movement of cargo from the outside to the inside of the cell. More specifically, we discovered the mechanism and the place where this defect manifests in early endosomes. This is the environment where all the cargo from the outside of the cell is sorted to be either recycled and released back to the outside of the cell, or where it is routed towards degradation in the lysosomes. In neurons, the regulation of endosomal trafficking is also a fundamental step that not only is central to synapse function, learning, and memory, but also the survival of the cell. All of these are impaired by ApoE4.
We have found that the ApoE4-mediated defect in endosomal trafficking and the resulting impaired synapse function are prevented by acidification of just the early endosomes. Depleting or inhibiting the cell of a protein called Sodium/Hydrogen Exchanger (NHE) 6 causes the acidification of early endosomes and alleviates the ApoE4-mediated endosomal trafficking defect (Xian X et al., 2018). Independent of the ApoE isoform, we also found that the ablation of NHE6 substantially reduces amyloid plaque formation, the initial pathological hallmark of AD in humans (Pohlkamp T et al., 2021). These conceptually novel and original findings suggest that increasing endosomal acidification with small molecules that can be taken by mouth could become an effective therapeutic approach to abolish the ApoE risk for AD and thus prevent the disease like the revolution by which the introduction of statins 30 years ago has reduced the incidence of myocardial infarction.
Featured on AlzForum, UT Southwestern News - 2018, and UT Southwestern News - 2021.
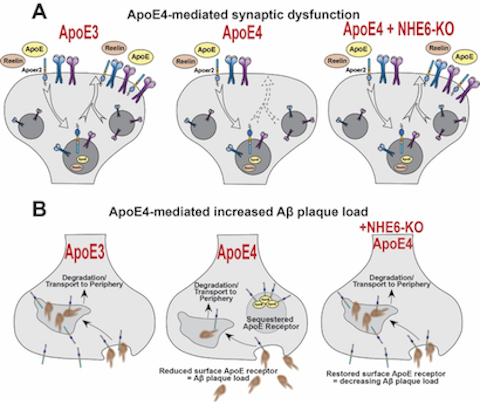 Therapeutic Potential of NHE6 as a target for Alzheimer's Disease
Therapeutic Potential of NHE6 as a target for Alzheimer's Disease Figure: Graphical summary of the therapeutic potential of NHE6 as a target for Alzheimer’s disease. (A) ApoE4 (middle), the highest risk factor for AD, causes synaptic dysfunction compared to the common ApoE3 allele (left). Synaptic dysfunction is restored upon genetic deletion of NHE6 (right, Xian X et al., 2018). (B) ApoE4 (middle) increases Aβ plaque deposition due to a reduction in Aβ plaque clearance compared to ApoE3 (left). Genetic ablation of NHE6 prevents and delays Aβ plaque formation in ApoE4 mice (right, Pohlkamp T et al., 2021).
Gene therapy for neurodegenerative diseases
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is another neurodegenerative disorder characterized by progressive dysfunction and loss of motor neurons. This ultimately results in muscle weakness and eventual paralysis with the first symptoms typically occurring in mid-adulthood. ALS is a rapidly progressing fatal disease and most patients die three to five years after diagnosis. Up to date, available treatment options are very limited and largely based on symptomatic therapies.
Although the exact mechanisms underlying the disease pathogenesis remain incompletely understood, it has been shown that excitotoxicity, neuroinflammatory processes, and disturbances in energy homeostasis are major contributors to neurodegeneration in ALS.
Considering the urgent need to identify new strategies to prevent neuronal death, we aim to study translational approaches to attenuate neurodegeneration and slow down disease progression in pre-clinical ALS models by targeting universal pathways involved in energy homeostasis and excitotoxicity as well as inflammatory responses in motor neurons.
Reelin Regulates Neuronal Excitability through Striatal-Enriched Protein Tyrosine Phosphatase (STEP61) and Calcium Permeable AMPARs in an NMDAR-Dependent Manner
Graphical Representation of Reelin Regulating Neuronal Excitability
Reelin is an important neuromodulator, which in the adult brain controls synaptic plasticity and protects against neurodegeneration. Amyloid-β has been shown to use mGluRs to induce synaptic depression through endocytosis of NMDA and AMPA receptors, a mechanism referred to as LTD, a paradigm of forgetfulness. Our studies showed that Reelin can block the internalization of these receptors through the regulation of tyrosine phosphatase STEP61, a protein that plays an important role in neurodegeneration. Moreover, Reelin deficiency increases the expression of calcium-permeable AMPA receptors in an NMDAR-dependent manner, which is pivotal to the formation of memories. Thus, Reelin uses mGluR pathways to regulate protein synthesis and its deficiency causes problems in memory formation as well as neurodegeneration.
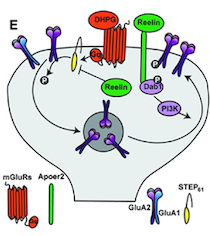 Graphical Representation of Reelin Regulating Neuronal Excitability
Graphical Representation of Reelin Regulating Neuronal Excitability The Role of the ApoE receptor-2 and the VLDL Receptor and their Ligand Reelin in Neuronal Migration, Axonal Transport, Neurotransmission, and Neurodegeneration
Emerging from the concept developed by my group that ApoE receptors serve as signal transducers and signal modulators in the vasculature and during brain development, we discovered pivotal new functions for ApoE receptors in the regulation of excitatory synapses (Weeber et al., 2002). There, ApoE receptors (Apoer2, Vldlr, LRP1 and LRP4) form complexes with glutamate receptors, tyrosine kinases, and other synaptic components to regulate the clustering, trafficking, and gating of ion channels (Beffert et al., 2005, Choi et al., 2013). These functions are differentially modulated by ApoE isoforms (Chen et al., 2010), glycosylation and proteolytic processing (Wasser et al., 2014). The insights that have emerged from this combined work now form a comprehensive basis on which the differential impact of ApoE isoforms and accumulating β-amyloid on synaptic function and dysfunction during the incipient stages of AD can be explained. They also offer conceptually novel treatment rationales that are based on the prevention or release of the endosomal recycling block induced by ApoE4.
The Role of Progranulin in Frontotemporal Dementia
Frontotemporal dementia (FTD) is the most common form of dementia in patients younger than 60 years of age. The socioeconomic burden of FTD is twice the amount per patient as Alzheimer’s disease and presents a major toll on our healthcare system as well as on patients and their families. Progranulin (PGRN) haploinsufficiency is a major genetic risk factor for developing FTD. PGRN is a ubiquitously expressed glycoprotein that consists of multiple granulin domains. Patients with homozygous PGRN mutations develop neuronal ceroid lipofuscinosis, a lysosomal storage disorder, implicating PGRN in lysosomal function. Preliminary data in mouse embryonic fibroblasts demonstrate massive alterations to the endo-lysosomal proteome dependent on PGRN gene dosage. However, how PGRN alters endo-lysosomal content and function remains unclear. Lysosomal and extracellular proteases cleave PGRN into multiple granulin domains, several of which have been shown to have effects distinct from those of full-length PGRN. This suggests these granulins are implicated in the various functions associated with PGRN. Recent advances in subcellular isolation methods such as “Lyso-IP” provide novel opportunities to study the effects of PGRN and its individual granulins on endo-lysosomal biology in neural systems in vitro and in vivo.
This project aims to investigate three key components of PGRN physiology: 1) the effect of Grn gene dosage on the endo-lysosomal proteome in vitro and in vivo, 2) the effect of individual granulin domains on endo-lysosomal composition, and 3) the necessity and sufficiency of individual granulins to ameliorate FTD-related pathologies in vivo. Answering these key questions will provide insight into the pathophysiology of several neurodegenerative diseases, which may open new therapeutic, preventative, or curative windows.
Evers BM, Rodriguez-Navas C, Tesla RJ, Prange-Kiel J, Wasser CR, Yoo KS, McDonald J, Cenik B, Ravenscroft TA, Plattner F, Rademakers R, Yu G, White CL and Herz J. (2017) Lipidomic and transcriptomic basis of lysosomal dysfunction in progranulin deficiency. Cell Reports 20, 2565-2574. PMID: 28903038.
The Role of LDL Receptor-Related Protein (LRP) in the Regulation of Smooth Muscle Cell Signaling, Vascular Remodeling, and Integrity in Atherosclerosis
The work from my group has shown that LDL receptor family members modulate a variety of fundamental cellular signaling pathways that participate in cellular cholesterol homeostasis in the vasculature and in the developing and adult brain, through proliferation, differentiation, and cell migration. Our early work on the role of LRP1 in the regulation of cellular cholesterol metabolism and export prompted us to investigate the role of LRP1 and other family members in the regulation of tissue remodeling (Bartelt et al., 2018) and inflammation (Jaoude et al., 2018, Ding et al., 2016) in detail. To this end, we and our collaborators generated tissue-specific knockouts for Reelin, LRP1, and the LRP1 adaptor protein Shc1 in the vascular wall. We further generated a knockin mutant that prevents the interaction of Shc1 with the tyrosine-phosphorylated intracellular domain of LRP1 (Jaoude et al., 2018). The results from these studies show crucial roles for LRP1 in the regulation of macrophage physiology, endothelial adhesion, and cholesterol metabolism, which revealed a novel mechanism underlying atherosclerosis (Ding et al., 2016, Xian et al., 2017).